



Gene Editing: The Key to Unlocking a Cystic Fibrosis Free World
A Glimpse into the Promising Frontier of Genetic Medicine for CF Patients


Imagine having a child knowing they only had forty years to live, a mere half of the average human lifespan. You watch them grapple with recurring bouts of pneumonia and intestinal blockages. They endure continuous assaults on their organs from numerous infections. Then, when they grow up, they face the relentless challenges of chronic respiratory failure and chronic bronchitis.
This is the harsh reality of cystic fibrosis, a debilitating condition that casts a shadow over the lives of more than 100,000 individuals worldwide. As of now, it is uncurable and the majority of patients only manage to reach their 30s or 40s.
However, hope is on the horizon. Over the next few decades, gene editing technologies promise to deliver a transformative, one-time curative treatment for cystic fibrosis by rectifying the CFTR gene mutations.
Cystic Fibrosis — What is it?
Cystic Fibrosis (CF) is a genetic disease caused by a mutation in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene.
A typical CFTR gene produces the CFTR protein responsible for transporting chloride ions in and out of cells. These ions play a pivotal role in regulating water movement, enabling the creation of low-viscosity mucus. This thin mucus serves as a protective barrier for the airway, digestive system, reproductive organs, and various other bodily tissues.

In the case of CF, an individual carries a mutated CFTR gene. This mutation leads to the production of abnormally thick and sticky mucus due to a lower amount of water. Consequently, this viscous mucus becomes the root cause of lung infections, wheezing, irregular bowel movements, and the emergence of salty sweat.

Over time, the accumulation of mucus in the lungs causes recurrent respiratory infections and progressive loss of lung function. Persistent inflammation and repeated lung damage often result in respiratory failure later in life, which is the most common cause of death from cystic fibrosis. Even though cystic fibrosis is a multiorgan disease that also affects the pancreas, liver, and other organs, mortality primarily results from respiratory failure.
Inheritance — How likely is CF in different types of patients?
CF is an autosomal recessive disorder. This means that both copies of the CFTR gene must have mutations in order for the disease to develop.
Everyone has two copies of each gene, one inherited from each parent. These are called alleles. In recessive conditions, both alleles must be faulty to cause disease.
If a person has only one faulty CFTR allele, they are known as a “carrier”. Carriers do not have cystic fibrosis, but they can pass the faulty gene to their children.
For a child to have CF, they must inherit one faulty CFTR allele from each parent. If both parents are carriers, there is a 25% chance with each pregnancy that their child will inherit CF.
Recessive disorders like CF are most common when both parents have a faulty gene copy. But they can also occur if only one parent is a carrier. In that case, the other faulty allele happens by chance due to a new mutation in the child.
Overall, understanding if parents are carriers through genetic testing is important to assess risk for their children. Both copies of the CFTR gene must be faulty to develop cystic fibrosis.

Testing
Diagnosing cystic fibrosis (CF) begins with a newborn screening, which detects elevated levels of immunoreactive trypsinogen (IRT).
If newborn screening is positive, it is followed up with a sweat test, which measures chloride levels in sweat. A sweat chloride level greater than 60 mmol/L indicates CF. However, intermediate sweat chloride levels (30–59 mmol/L) require genetic testing for confirmation.
Molecular genetic testing of the CFTR gene identifies mutations on both alleles, confirming the diagnosis of CF. Over 1000 CFTR mutations have been identified, but a panel of the most common mutations is typically tested first.
If only one or no mutations are found, full gene sequencing of the CFTR gene may be conducted to identify rarer mutations. Identifying the specific CFTR mutations in a patient is important to guide prognosis and treatment decisions.
Other supportive tests include chest radiography to assess lung disease, sputum culture to identify lung infections, and fecal elastase testing to assess pancreatic involvement. Regular monitoring of lung function with spirometry is a key part of long-term management.
Current CF Treatments
Many treatments currently exist for CF. They may contain medications, exercises or special therapy vests and aim to control symptoms, even though Cystic Fibrosis is untreatable.
Symptom Medications
Antibiotics: These are commonly used to prevent and treat infections that can affect the lungs and other organs in CF patients.
Mucus Thinners/Reducers: Medications that help thin or reduce the thick and sticky mucus in the airways, making it easier to clear and less destructive.
Bronchodilators: These medications help widen the airways, making it easier for individuals with CF to breathe and improve airflow.
Steroid Medication: In some cases, steroids may be prescribed to manage inflammation and control the growth of abnormal tissue or polyps in the airways.
Lung Transplantation
For some individuals with severe lung disease, lung transplantation may be considered as a treatment option. This does not cure the underlying genetic defect in CF, but it can provide healthy lungs that are not affected by the disease. Lung transplants for CF patients have become more successful in recent years. However, it remains a major procedure with significant risks of complications.
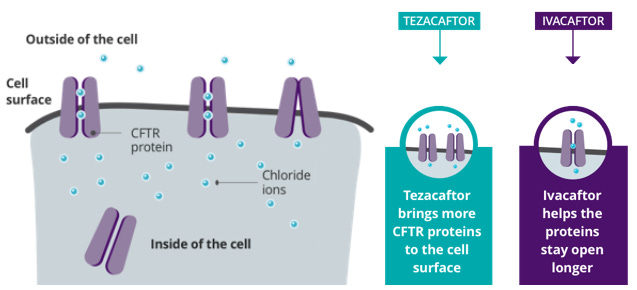
CFTR Protein Modulators
In specific mutations, people may be able to use special drugs that help correct and restore function within the CFTR protein created by the faulty CFTR gene. This is not gene editing, as it only changes the protein that is created by the gene.
Most modulators are a combination of medications, with Ivacaftor being the most commonly utilized. Ivacaftor is a potentiator that attaches to the faulty protein on the cell surface and opens the chloride channel and gate to allow chloride ions to pass through.
Lumacaftor, elexacaftor and tezacaftor are all correctors that help change the shape of CFTR proteins so that they float to the surface of a cell. It essentially brings more proteins to allow for more chloride ion flow.
Airway Clearance Techniques
Breathing techniques like Active Cycle of Breathing Techniques (ACBT) involve controlled breathing exercises such as pursed-lip breathing, small and deep breaths, breath holds, and forced exhalations (huffs/huff coughs) to open airways and mobilize mucus.
Physical chest therapies like percussion, positioning, vibration aids, and simulated coughing devices help loosen, mobilize, and drain mucus. Examples include chest percussion, positioning the body to use gravity, hand-held vibrators, and mechanical cough simulators.
Specialized devices like high-frequency chest wall oscillation vests automatically shake the chest wall rapidly to loosen mucus for easier clearance. Autogenic drainage and modified postural drainage also use breathing control and gravity through positioning to clear mucus from the lungs.

CF management extends beyond medications and airway clearance techniques. Nutritional counseling, physical exercise, and adherence to specialized diets prescribed by healthcare providers can significantly reduce the impact of CF on an individual’s health and well-being.
Genetic Engineering Solutions and Breakthroughs
Genetic Engineering and Gene Editing. What’s the difference?
Gene editing refers to directly modifying DNA sequences in a precise way using tools like CRISPR. This allows intentional changes to be made without introducing foreign DNA.
Genetic engineering more broadly refers to introducing new DNA sequences, often from other species, to give an organism new traits. This can be done through various techniques like direct DNA injection or using viral vectors.
While the techniques differ, current CF therapies in development use both approaches. 4D-710 and SPL84 utilize gene editing to directly correct mutations. LUNAR-CF and VX-522 employ genetic engineering to deliver mRNA that provides instructions to produce normal CFTR protein without fixing underlying DNA.
Current Clinical Trials & Promising Research
Currently, numerous genetic engineering and editing drugs are being developed for CF patients. However, all are still in early clinical trials — phase 2 or lower. The therapies in development take different approaches to potentially restore CFTR function by delivering genes, mRNA, or splicing factors directly to lung cells. While the techniques vary, the goal is the same — provide cells with what they need to make normal CFTR protein, regardless of the underlying genetic mutations.
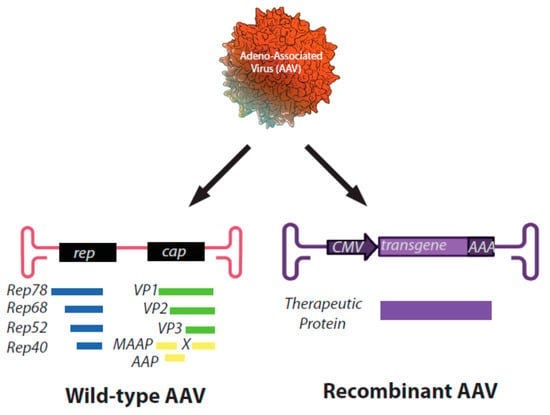
4D-710: 4D-710 uses an engineered adeno-associated virus (AAV) as a vector to deliver normal CFTR genes directly to lung cells. The AAV injects DNA containing functioning CFTR genes into the cells. The cells can then use these genes to produce normal CFTR protein, regardless of the underlying CFTR mutation. It is also an inhaled aerosol.
AAVs or Adeno-associated viral vectors are a way to deliver genes. They are viruses that have been changed, so they can carry new genes into human cells safely.
The outer shell of the virus is altered, so it doesn’t cause infection, but can still enter cells
The original inner genes are removed and replaced with normal CFTR genes
When injected, the viral vector goes into cells and releases the CFTR gene. The new gene can then start producing the CFTR protein properly. Adeno-associated viral vectors are a promising approach for gene therapy because they work well and don’t seem to cause major side effects.
SPL84: SPL84 is a small genetic fragment designed to fix splicing mutations — errors in cutting and reassembling RNA strands. It works by binding to the RNA and modifying how the strands are spliced together. This allows the RNA to be assembled properly, enabling the production of normal CFTR protein despite splicing mutations in the DNA. SPL84 is delivered to your lungs by inhalation, where it can enter the cells and reach their nuclei, where splicing happens.
The specific mutation it corrects is called 3849+10 kb C->T, and it affects how the gene is turned into protein. To make a protein, the gene first needs to be copied into RNA, which carries the information from the gene to create proteins. The RNA is made of two types of parts: exons, which are useful and contain the information for the protein, and introns, which need to be removed.
The process of removing the introns from the RNA is called splicing, and it is done by a spliceosome. The spliceosome can recognize which parts are exons and which parts are introns by looking for certain signals in the RNA. However, in the case of the 3849+10 kb C->T mutation, one of these signals is changed. The intron is interpreted as an exon, and it includes it in the final RNA. This extra part is called a cryptic exon, and it causes issues. As a result, most of the RNA is destroyed by a system called Nonsense mediated mRNA decay (NMD), and the remaining RNA makes a short and broken protein that does not work well.
SPL84 is a new treatment for CF that can fix this splicing problem. It is a short synthetic molecule that can attach itself to the RNA near the cryptic exon. By doing so, it covers up the signal that makes confuses the spliceosome. This way, it restores the normal splicing pattern, and makes sure that only the exons are included in the final RNA. This leads to the production of a normal and functional CFTR protein that can help regulate the salt and water balance in your body.
LUNAR®-CF: LUNAR®-CF skips over the defective CFTR DNA entirely and instead delivers mRNA — a temporary messenger copy of the genetic code — directly into lung cells. The mRNA provides the cells with instructions to produce fully functional CFTR protein. This mRNA therapy could potentially benefit all people with CF, not just specific mutations. This will be inhaled.
LUNAR stands for Lipid-enabled and Unlocked Nucleomonomer Agent modified RNA. It is a delivery system developed by Arcturus Therapeutics to safely and effectively deliver mRNA therapeutics to target cells.
The key components of LUNAR are:
The mRNA drug — This is the therapeutic mRNA encoding the desired protein, in this case CFTR mRNA to fix the defect causing cystic fibrosis.
Lipids — These are fatty molecules that protect the mRNA and help it get inside cells. The lipid coating helps the mRNA evade enzymes that would break it down before delivery.
Proprietary modifications — The mRNA is chemically modified in a proprietary way to improve stability and prevent the immune system from recognizing it as foreign.
The mRNA and lipids self-assemble into nanoparticles that can be inhaled directly into the lungs. The particles are designed to fuse with lung cell membranes and release the mRNA drug into lung cells. Once inside cells, the mRNA produces the desired protein — in this case CFTR protein to restore chloride transport in cystic fibrosis patients.
The key advantages of LUNAR are its ability to deliver mRNA efficiently into lung cells and its biocompatibility for inhaled therapeutics. If proven safe and effective, it could allow mRNA therapies to reach previously untreatable lung diseases.
VX-522 mRNA: Similar to LUNAR®-CF, VX-522 delivers mRNA via lipid nanoparticles directly into lung cells. This provides the instructions to produce normal CFTR protein without needing to correct the underlying DNA. As an mRNA therapy, it has the potential to help all individuals with CF. This will be inhaled and is similar to LUNAR®-CF in terms of technique.
Ethics
Gene Editing may cure Cystic Fibrosis one day, but the ethicality of each aspect must be considered to ensure that everyone is safe.
As gene editing advances, a few things that must be considered include:
Testing extensively for safety. Study all effects, even rare or delayed ones, to ensure no unintended harm.
Ensuring fair access. Treatments should be available to all who need them, not just those who can pay.
Proceeding with compassion. Use this technology thoughtfully to relieve suffering and better human lives.
The Outlook Ahead
While cystic fibrosis remains a challenging condition, the future is brighter than ever thanks to recent advances and future possibilities.
Gene editing has the potential to correct the underlying genetic mutations that cause CF. If proven safe and effective through clinical trials, such direct gene editing treatments could be truly transformative — offering the possibility of a permanent genetic cure.
If our society continues to fund and support research, cystic fibrosis may one day become a very manageable condition rather than the life-threatening illness it has often been.
In summary, while challenges persist, the future is brighter than ever for those with cystic fibrosis thanks to dedicated researchers. Each breakthrough brings us one step closer to a world free of this disease.
References
(n.d.). Cystic Fibrosis Canada. Retrieved October 26, 2023, from
https://www.cysticfibrosis.ca/
‘;;’. (2019, March 9). ‘;;’ — YouTube. Retrieved October 26, 2023, from https://apps.cff.org/Trials/Pipeline/details/10208/SPL84
‘;;’. (2019, March 9). ‘;;’ — YouTube. Retrieved October 26, 2023, from https://apps.cff.org/Trials/Pipeline/details/10204/VX-522-mRNA
About Cystic Fibrosis. (n.d.). Cystic Fibrosis Foundation. Retrieved October 26, 2023, from https://www.cff.org/intro-cf/about-cystic-fibrosis
About Us. (n.d.). Cystic Fibrosis Canada. Retrieved October 26, 2023, from https://www.cysticfibrosis.ca/about-us/cf-ambassadors/c%C3%A9line-dion
Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. (2017, July 1). NCBI. Retrieved October 25, 2023, from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5548848/
Arcturus’ Pipeline of mRNA Medicines and Vaccines for Rare Diseases Cystic Fibrosis (CF), Covid-19, Influenza (Flu), Ornithine Transcarbamylase Deficiency (OTCD). (n.d.). Arcturus Therapeutics. Retrieved October 25, 2023, from https://arcturusrx.com/mrna-medicines-pipeline/#lunarCf
Bower, J. J. (n.d.). Harnessing the Natural Biology of Adeno-Associated Virus to Enhance the Efficacy of Cancer Gene Therapy. MDPI. Retrieved October 26, 2023, from https://www.mdpi.com/1999-4915/13/7/1205
Cystic Fibrosis (CF): Causes, Symptoms, Diagnosis & Treatment. (2021, October 8). Cleveland Clinic. Retrieved October 26, 2023, from https://my.clevelandclinic.org/health/diseases/9358-cystic-fibrosis
Cystic fibrosis — Symptoms and causes. (2021, November 23). Mayo Clinic. Retrieved October 26, 2023, from https://www.mayoclinic.org/diseases-conditions/cystic-fibrosis/symptoms-causes/syc-20353700
Delivery Characterization of SPL84 Inhaled Antisense Oligonucleotide Drug for 3849 + 10 kb C- > T Cystic Fibrosis Patients. (n.d.). PubMed. Retrieved October 25, 2023, from https://pubmed.ncbi.nlm.nih.gov/37643307/
Drug Development Pipeline | CFF Clinical Trials Tool. (n.d.). Find a CF Care Center. Retrieved October 26, 2023, from https://apps.cff.org/trials/pipeline
4D-710 by 4D Molecular Therapeutics for Cystic Fibrosis: Likelihood of Approval. (2023, September 11). Pharmaceutical Technology. Retrieved October 25, 2023, from https://www.pharmaceutical-technology.com/data-insights/4d-710-4d-molecular-therapeutics-cystic-fibrosis-likelihood-of-approval/
4D-710 | CFF Clinical Trials Tool. (n.d.). Find a CF Care Center. Retrieved October 26, 2023, from https://apps.cff.org/Trials/Pipeline/details/10197/4D-710
LUNAR®-CF | CFF Clinical Trials Tool. (n.d.). Find a CF Care Center. Retrieved October 26, 2023, from https://apps.cff.org/Trials/Pipeline/details/10159/LUNAR-CF
Newborn Screening for CF. (n.d.). Cystic Fibrosis Foundation. Retrieved October 26, 2023, from https://www.cff.org/intro-cf/newborn-screening-cf
Pagaduan, J. V. (2020, June 5). Immunoreactive Trypsinogen (IRT). Testing.com. Retrieved October 26, 2023, from https://www.testing.com/tests/immunoreactive-trypsinogen-irt/
RNA Vaccines | Lipid-Mediated Delivery | mRNA Technology. (n.d.). Arcturus Therapeutics. Retrieved October 25, 2023, from https://arcturusrx.com/rna-mrna-proprietary-technologies/
Testing for CF. (n.d.). Cystic Fibrosis Foundation. Retrieved October 26, 2023, from https://www.cff.org/intro-cf/testing-cf
Great summary of CF and the treatment options currently available and on the horizon. It gives me hope that my daughters may not have to live with CF forever. One thing you didn't mentiría that carriers can also exhibit CF symptoms without having CF. They're currently calling it CFTR Related Syndrome. It is my situation. I didn't know I was a carrier until my daughters were diagnosed. I was tested and don't have two bad genes, just the one. But I have pancreatic insufficiency and Covid seemed to activate something in my lungs and now I require daily therapy to keep my lungs clean. The challenge is that CFTR Related Disorder is not recognized by insurers and so I'm not eligible for any of the medications like Pulmozyne and Trikafta that might help me more than sodium chloride and a therapy jacket.
Very informative, Jonathan!